Everyday, Millions of customers usually shopping on Amazon.com. If you are an Amazon’s customer, let spend 5 minutes to check the latest Amazon deals week coupon code 2013 and buy many products at very attractive price.
Amazon deals week coupon code 2013
Click to see live deals : Thousands of products discount up to 50%, 75% off

View some small coupons on Amazon.com (May be used for exact products)

Use this coupon: 30OFF100 to save 30% off when spend more than $100, use it to buy : Handbags, shoes, clothings
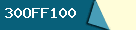
Use coupon: SWTRCSHM to get 20% off for Sweaters, Cashmere ... for men, women etc, click to activate and see more detail

Click and see featured deals and discounts for Electronics and accessories today

View all deals and coupon for HDTV, 3D, Smart TV from LED, Plasma by Samsung, LG, Sony, Panasonic, Vizio all sizes and accessories
View the latest coupon, deals for cameras from Nikon, Canon, Sony, Panasonic, Samsung and accessories
Laptops, ultrabooks from Dell, Toshiba, Samsung, HP, Asus, Apple, Acer discount up to $200, 20% etc
Click to show all Shoes coupon, deals and sales on Amazon.com at this time
Click to see all watches discounts, deals today: up to 20%, 30% off for most popular brands: Seiko, Timex, Casio, Rolex etc
Read some buying guide below:
Amazon deals week coupon code 2013 usually available for : Baby, Shoes, Sandal, Handbag, Clothing, Wallet. With another departments such as: Electronics (HDTV, Camera, Laptop, Phone and accessories), Books, Games, Toys, Watches, Jewelry or Automotive, you can’t find coupon or promo code for it. Amazon usually offers: Deals, sales and special offers or discount directly. We are ready for the latest Amazon deals week coupon code 2013, Amazon promo code 2013.

Everyone knows that Amazon is one of the biggest as well as the widest shopping portals in the world. Since it is one of the biggest shopping platforms in the world, it is no wonder that most people would go shopping there. Although there are also other shopping portals that are offering new and sophisticated ways in shopping, people are always go back to Amazon for more. So, what are the reasons to shop on Amazon, anyway?
Amazon.com encourages the use of coupons while shopping on the website. It is one of the few shopping sites people are attracted to for its coupons deals and discounts. However, it is always wise to make considerations before using Amazon coupon code 2013 (Amazon promo code 2013 – Amazon discount codes 2013) to maximize the advantage attached to them. Some of them include free shipping and discounted merchandise.